
Autoria: Margarida Almeida (MEBiol)
O Prémio Nobel da Química de 2024 foi dividido entre David Baker que computacionalmente consegue criar proteínas das quais a natureza ainda não se lembrou, e Demis Hassabis e John Jumper que desenvolveram um modelo de inteligência artificial, o AlphaFold2, que prevê a estrutura tridimensional de proteínas. O Diferencial entrevistou Ângela Taipa, professora e investigadora no Instituto Superior Técnico, para comentar a atribuição deste prémio.
A 9 de Novembro foi publicado o comunicado à imprensa, por parte da Academia Real das Ciências da Suécia, que anunciava que o Prémio Nobel da Química de 2024 seria dividido em duas partes, sendo a primeira metade atribuída a David Baker, pelo design computacional de novas proteínas artificiais, e a segunda, a Demis Hassabis e John Jumper pelo desenvolvimento de um modelo de aprendizagem para a previsão de estruturas proteicas.
Sobre proteínas
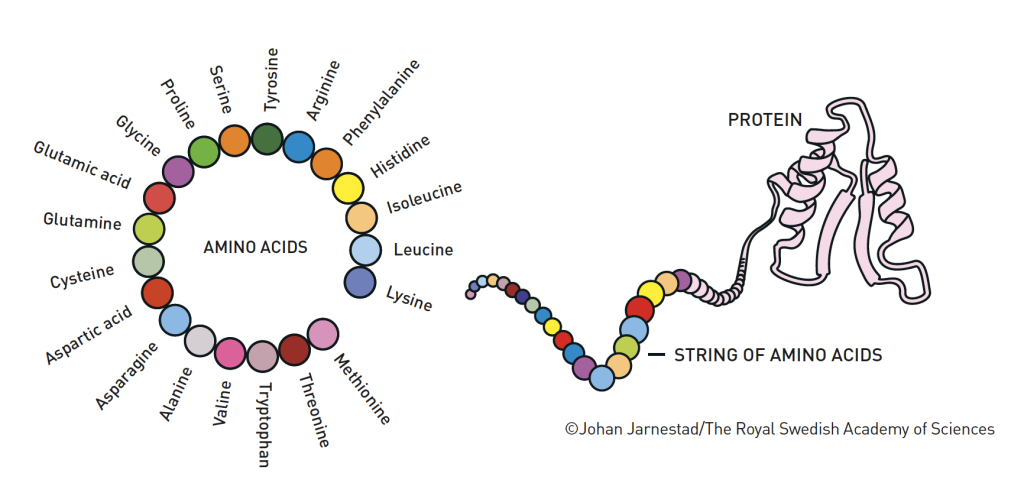
As proteínas são sequências de aminoácidos que resultam da expressão de genes. O código genético é a nossa receita e as proteínas, os ingredientes. A receita traduz-se em ingredientes que devem ser obtidos para depois serem utilizados na manutenção da nossa saúde. Todas as funções do nosso corpo decorrem da síntese de proteínas.
Temos 20 aminoácidos essenciais, as unidades de que são feitas as proteínas, que são cadeias de grande dimensão que se enrolam tridimensionalmente sobre si de forma a alcançar o estado energeticamente mais estável, sendo esta estrutura tridimensional responsável pela função da proteína.
Cada proteína distingue-se quimicamente das restantes pela sua sequência de aminoácidos. Teoricamente, uma mesma sequência de aminoácidos origina estruturas tridimensionais semelhantes.
A Primeira Metade
David Baker ganhou proeminência em 2003 com a publicação de um artigo na revista Science sobre o processo de criação da proteína Top 7, uma nova proteína não existente na natureza.
Processo esse que passou pela determinação da potencial estrutura da proteína com base na funcionalidade desejada e conhecimento disponível sobre subunidades proteicas, pelo cálculo de mínimos de energia para essa estrutura, pela substituição de aminoácidos “em posições específicas para alcançar energias progressivamente mais pequenas, até chegar a uma sequência de aminoácidos de energia mínima”. Tudo isto executado de forma computacional recorrendo ao algoritmo Rosetta cuja criação e desenvolvimento se atribui principalmente a Baker.

“É o contrário do paradigma de sempre que era tentar perceber qual a estrutura de uma proteína e a sua função a partir da sua sequência de aminoácidos. E o que ele faz é o processo inverso”, comenta Ângela Taipa, professora do departamento de Bioengenharia (DBE) do Instituto Superior Técnico e investigadora no Instituto de Bioengenharia e Biociências (iBB).
Anteriormente a esta publicação, os estudos na área de Design de Proteínas dedicavam-se primariamente à imitação ou optimização de estruturas proteicas existentes. O que a equipa de Baker trata é a construção de raíz de uma nova proteína.
Introduzindo o gene que codifica para a sequência de aminoácidos proposta pelo algoritmo numa bactéria, este é expresso, a proteína desenhada é produzida e purificada, podendo a sua estrutura ser verificada por métodos experimentais e comparada com a estrutura determinada durante o processo de desenvolvimento.
No caso da Top 7, a estrutura verificada por cristalografia de raios-X revelou-se muito próxima àquela que tinha sido proposta pelo algoritmo. Desde então, Baker tem produzido várias outras ‘criações’.
No entanto, a professora refere: “Aqui fala-se essencialmente em estabilidade, mas se a estrutura tridimensional da nova proteína é idêntica à determinada experimentalmente, em princípio, a proteína resultante terá a atividade biológica para a qual foi desenhada, o que pode ser verificado.”
A Segunda Metade
Demis Hassabis e John Jumper desenvolveram um robusto modelo de inteligência artificial, o AlphaFold2, para a previsão da estrutura tridimensional de proteínas dada uma sequência de aminoácidos.
Este modelo foi desenvolvido a partir do treino de algoritmos de aprendizagem computacional com pares de sequências de aminoácidos e respectivas estruturas tridimensionais proteicas experimentalmente determinadas.
O processo de determinação experimental da estrutura tridimensional de uma proteína é dispendioso e extremamente lento, podendo demorar meses ou anos.
Um método popular para este fim é a já referida cristalografia de raios-X que envolve a colocação de uma proteína perante um feixe de raios-X. Conforme a interferência causada, a estrutura cristalina da proteína é determinada. O desenvolvimento desta metodologia valeu, em 1961, um prémio Nobel a John Kendrew e a Max Perutz. Desde então, foram determinadas as estruturas cristalinas de milhares de proteínas, tendo sido esses dados acumulados que permitiram o treino deste modelo e o funcionamento do Rosetta que pesquisa as bases de dados de estruturas proteicas de modo a produzir o seu output.
Originalmente, o algoritmo de Baker não utilizava inteligência artificial, mas após observar o sucesso de Hassabis e Jumper, acabou por decidir também potenciar o Rosetta com essa abordagem.
O Futuro
Existem mais de 100.000 proteínas diferentes apenas no corpo humano, pelo que o desenvolvimento de métodos que permitam acelerar e sistematizar os estudos biológicos é determinante. Este tipo de ferramentas vem ainda potenciar a criatividade dos investigadores para propor abordagens inovadoras ao nível da biologia molecular para a resolução dos mais variados problemas.
David Baker repete em várias das suas apresentações: «A natureza faria o seu papel como fez ao longo de milhões de anos. Iria mutar proteínas que nos permitiriam resolver alguns [desses] problemas, mas o processo é muito lento.»,
Estas expansões podem vir a revolucionar o conhecimento relativo aos acontecimentos moleculares envolvidos na evolução de doenças, na resistência a medicações e a terapias, no desenvolvimento humano e na expressão de funções moleculares desejáveis.
O Futuro (para os mais fluentes em Biologia Molecular)
Um dos próximos desafios a que o laboratório de Baker se propõe será o desenvolvimento de molecular machines, “cascatas de proteínas que atuam […] essencialmente nas células. Alimentam-se de um fuel, que normalmente é ATP ou ADP ou outra molécula deste género e eliminam ‘junk material’. […] O que é que ele quer? Quer desenhar molecular machines para pôr em circulação e eliminar algum lixo celular, por exemplo, compostos que estejam na base da formação de agregados beta-amiloides que são responsáveis por algumas doenças neurodegenerativas.”
Relativamente à expansão do trabalho de Hassabis e Jumper, a professora faz a seguinte observação: “Há cerca de 15, 20 anos, descobriu-se uma nova classe de proteínas, as proteínas intrinsecamente desordenadas”, ou intrinsically disordered proteins (IDPs). “Proteínas que têm zonas totalmente desordenadas, mas que têm atividade biológica. E isso não está previsto no paradigma sequência de aminoácidos-estrutura-função e os padrões de folding não têm em conta estas partes totalmente desordenadas da proteína que são complexas. Há uma série destas proteínas que estão relacionadas com doenças degenerativas. Será um grande desafio arranjar um AlphaFold2 para prever a estrutura destas proteínas, porque a variabilidade em termos de composição de aminoácidos é muito maior.”
Segundo a revista Science, o AlphaFold3, lançado em 2021, vai além do seu antecessor na medida em que consegue prever, não só a estrutura tridimensional das proteínas, mas também como estas se ligam e interagem com as outras moléculas, incluindo o DNA e o RNA.
Notas finais
Para contextualização histórica e científica do Prémio Nobel da Química, o Comité Nobel disponibiliza os seguintes documentos consultados durante a elaboração deste artigo: “They have revealed proteins’ secrets through computing and artificial intelligence” e “Computational protein design and protein structure prediction” para informação avançada.
